

#apaperaday: Systemic PPMO-mediated dystrophin expression in the Dup2 mouse model of Duchenne muscular dystrophy
In today’s #apaperaday, Prof. Aartsma-Rus reads and comments on the paper titled: Systemic PPMO-mediated dystrophin expression in the Dup2 mouse model of Duchenne muscular dystrophy
The LUMC Leiden with a paper from Molecular Therapy Nucleic Acids by Gushchina et al from the Wein / Kevin Flanigan team on dystrophin exon 2 skipping in the exon 2 duplication mouse model using antisense oligonucleotides. Doi 10.1016/j.omtn.2022.10.025.
Duplications of exon 2 are the most common of duplications in Duchenne. Exon skipping for duplications is challenging as for multi-exon duplications you need to target the right exon but half the time the wrong one will be targeted and the reading frame will not be restored.
For single exon duplications, skipping either exon will restore normal transcripts. HOWEVER, skipping both, will disrupt it. Exon 2 is an exception however. Here if you skip 2 exons, you get an exon 2 deletion, which still allows dystrophin production via an IRES in exon 5
Authors already showed exon 2 skipping is feasible with an antisense gene (U7snRNA delivered with AAV) in their exon 2 duplication mouse and in 3 patients. Here they tested peptide conjugated phosphorodiamidate oligomers (PPMOs) targeting exon 2 as an alternative.
PPMOs are antisense oligonucleotides (ASO). Usually PMOs are 25mers. However, the authors here use a 31mer. This is very long. Having worked with exon 2, I know it is an ASO design nightmare (very high A content, so difficult to get a good binding ASO).
This may explain the long length – authors do not elaborate about this though. They used systemic treatment of their exon 2 duplication mouse with 20, 40 or 80 mg/kg and saw exon 2 single and double skipping in a dose dependent manner in all skeletal muscle and less in heart.
They looked at different days post injections and saw highest exon skipping between 7-15 days, after which exon skipping levels reduced but were still detectable after 60 days in skeletal muscle.
They also looked at dystrophin restoration showing that ~80% of fibers became dystrophin positive for skeletal muscles (~7%). Western blotting showed ~20% dystrophin levels in skeletal muscle, with higher levels in diaphragm and lower in heart.
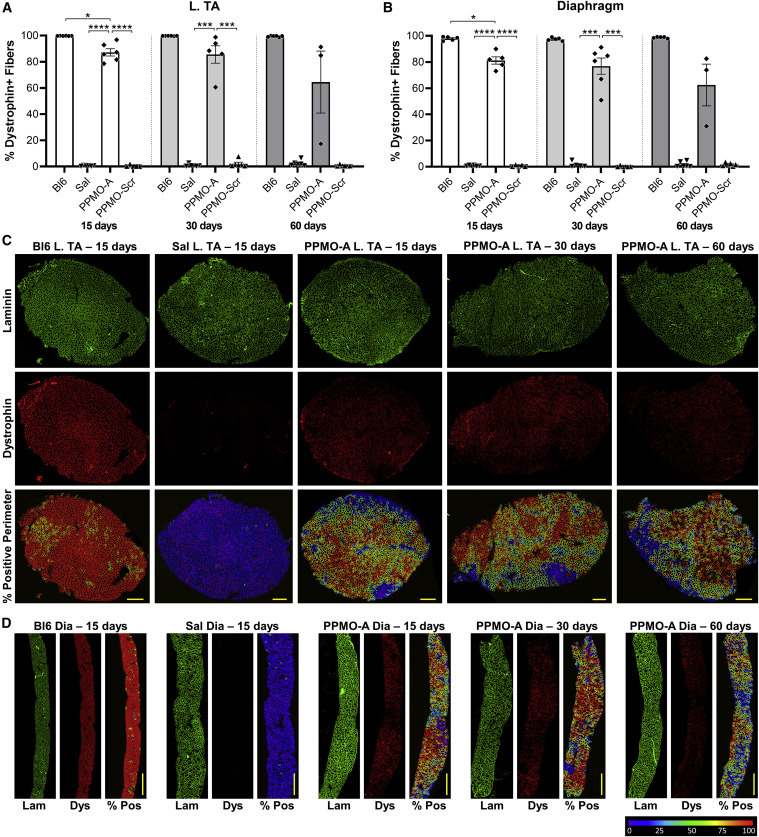
Finally authors looked at centrally located nuclei levels, which were reduced in treated vs untreated mice in the TA and the diaphragm. Many good things: authors looked into different muscles, had controls and looked at RNA and protein and some histological parameters.
However, they did not look into fibrosis, general histology or muscle function yet. Authors discuss more work is needed to optimize dosing and regimen – I agree and also understand that more labor intensive studies like muscle function are postponed until optimization is over
Authors discuss why they developed exon 2 skipping PPMOs when they already have AAV exon 2 antisense gene operational: 1. Not all patients can be treated with AAV due to preexisting immunity 2. It is difficult to produce AAV at large scale for clinical use as an academic group.
They use a PPMO from Sarepta, but it is not mentioned whether the peptide is the same as in vesleteplirsen. Authors state that they expect the exon 2 skip dystrophin to be more functional than microdystrophin – i agree, especially when 1 exon 2 is skipped (normal dystrophin)
They outline that PPMO treatment may have less risks for side effects than AAV. This may be the case, but the arginine rich peptides have a risk for kidney toxicity that is dose limiting. The dose that is needed to get toxicity appears to be lower in humans than mice.
So the fact that the doses used in mice are well tolerated in mice (up to 80 mg/kg) does not mean they will be tolerable in humans. More work is needed as the authors indicate. Still, nice early work with beautiful immunofluorescence images!

