

#apaperaday: Cell-mediated exon skipping normalizes dystrophin expression and muscle function in a new mouse model of Duchenne Muscular Dystrophy
In today’s #apaperaday, Prof. Aartsma-Rus reads and comments on the paper titled: Cell-mediated exon skipping normalizes dystrophin expression and muscle function in a new mouse model of Duchenne Muscular Dystrophy
From home because I am a bit under the weather (disclaimer) and Yuzu wanted to explore the mobile rather than pose. Today’s pick is from Galli et al on cell-mediated exon skipping in a mouse model from @EmboMolMed DOI: 10.1038/s44321-024-00031-3
Duchenne is caused by lack of functional dystrophin, which makes muscle fibers sensitive to contraction-induced damage and chronic muscle pathology. Becker patients have partially functional dystrophin and have a later onset and slower disease, but also muscle pathology.
Authors here want to pursue cell therapy as they argue that muscle fibers persist life long so if successful this would result in a long term treatment effect. Some counter arguments from me:
- Authors here use cells with a partially functional dystrophin, so the muscle fibers will not persist life long (as we know from Becker).
- The cells will go into a pathological environment which is not the same as healthy muscle.
Back to the paper: authors use autologous cells & add an exon skip gene (U7 snRNP) to skip exon 51. They use a lentiviral vector to deliver the exon skipping gene to immortalized myoblasts (and confirm results in transdifferentiated fibroblasts and primary mesangioblasts).
Authors give the viral vector also a green signal so they can identify transduced cells from a patient with an exon 51 skippable mutation. They confirmed exon 51 skipping in the cells with the U7 snRNP and dystrophin restoration with immune fluorescence
Authors speculate that the U7 snRNP might migrate between muscle cells and therefore they perform co-cultures of the patient cells with also the U7 snRNP containing patient cells in ratios of 1:10 and 1:30, arguing this is similar to the delivery efficiency in patients (0.7%)
Note that there are many differences between cultured cells at a ratio (where some cells may proliferate quicker than others and they fuse with each other) and a human muscle with severe pathology where the transplanted cells have to fuse with damaged muscle fibers
What is interesting is that authors indeed see that the U7 snRNP migrates between cells and that there is more dystrophin produced than expected from a 1 in 10 dilution and also more than from a 1 in 10 dilution of wild type and patient cells.
Authors show this with western blot – sadly they do not use a concentration series but quantify compared to only one wild type sample and correct for myosin (see image). As mentioned authors tested this also with primary mesangioblasts and fibroblasts myoDed into myotubes.
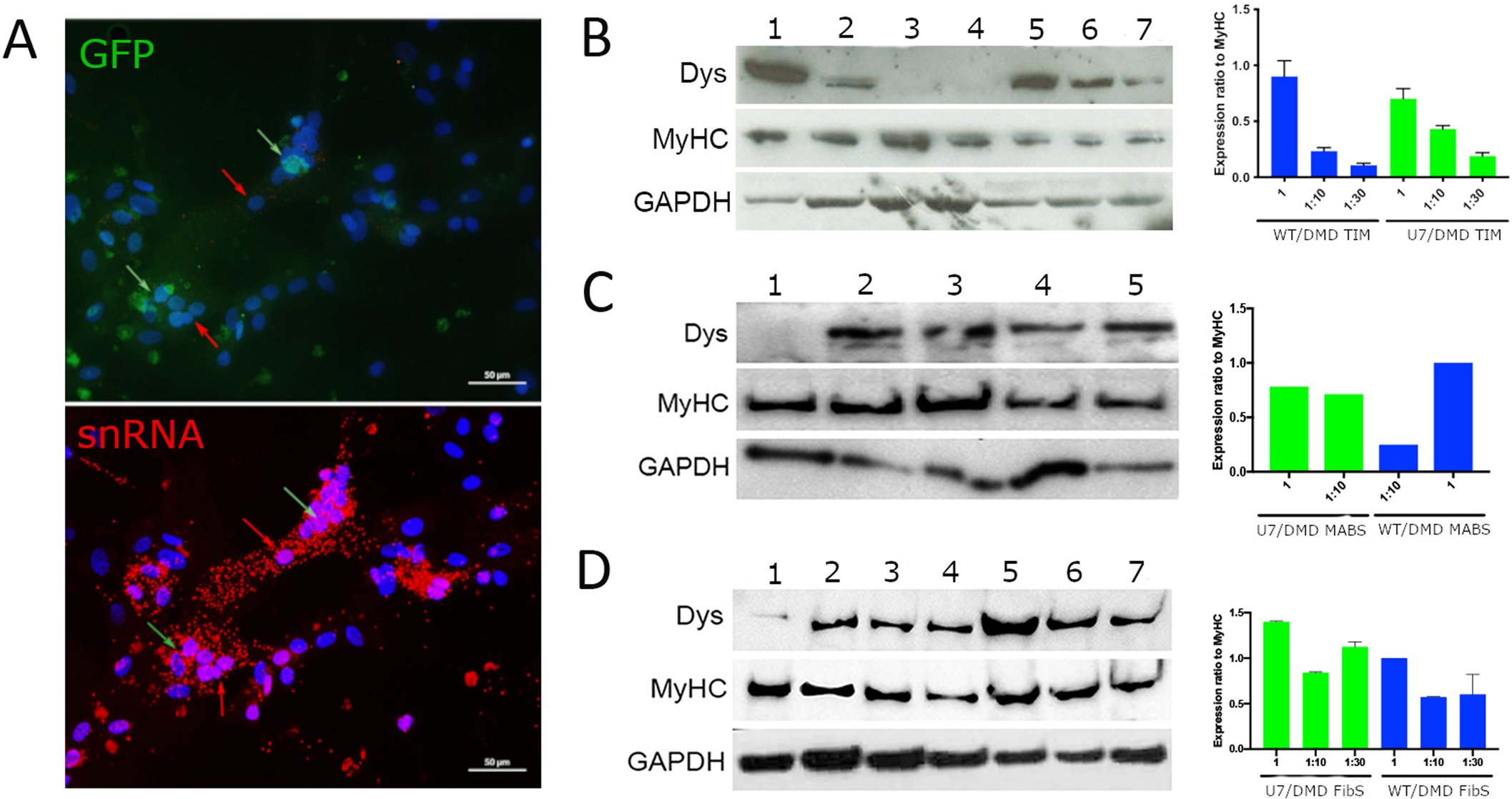
They report the same trend. Then they tested the immortalized myoblasts in a mouse model. To this end they generated a deletion that can be skipped by exon 51 skipping, and they humanized the splice site target sequences of the U7 snRNP system.
The mouse model is described with little detail – it is on an immune deficient background (needed to transplant human cells). Note a humanized model that needs exon 51 skipping is available already – we share this under an MTA with academic collaborators.
A dystrophic Duchenne mouse model for testing human antisense oligonucleotides
I am pointing this out as probably getting our mouse model and crossing it to an immune deficient background would have cost less mice than making a new line (but difficult to discern due to the paucity of information in the methods)
Back to the paper: authors perform intramuscular injections of the human cells that were transduced and detect dystrophin restoration. When doing a dose decrease they see that the level of decrease is larger than the decrease in dystrophin produced.
So in other words: it seems the U7 snRNP migrates also after intramuscular injection in a mouse muscle (which has very active regeneration so we should be careful with extrapolation to the human system). Authors show dystrophin persists for at least 11 months.
Authors performed a motility assay in mice treated locally in gastrocnemius, TA and quadriceps, showing increased motility. There is little information on the motility assay but treated mice do as good as wild types (not expected as they produce partially functional dystrophin)
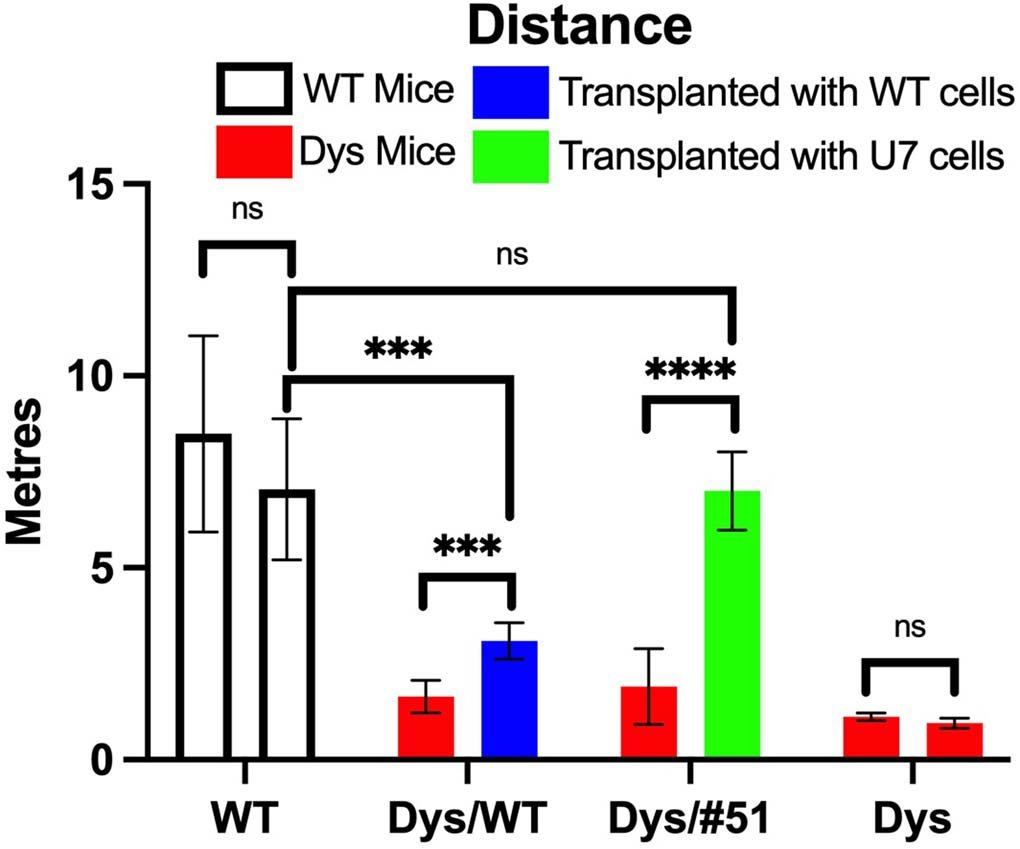
Also if I interpret the test & graph correctly mice run only 10 meters in 15 minutes while the speed was at 15 meters per minute (???). Finally authors performed intraarterial injections and saw low levels of dystrophin restoration – now corrected by GAPDH rather than myosin (?)
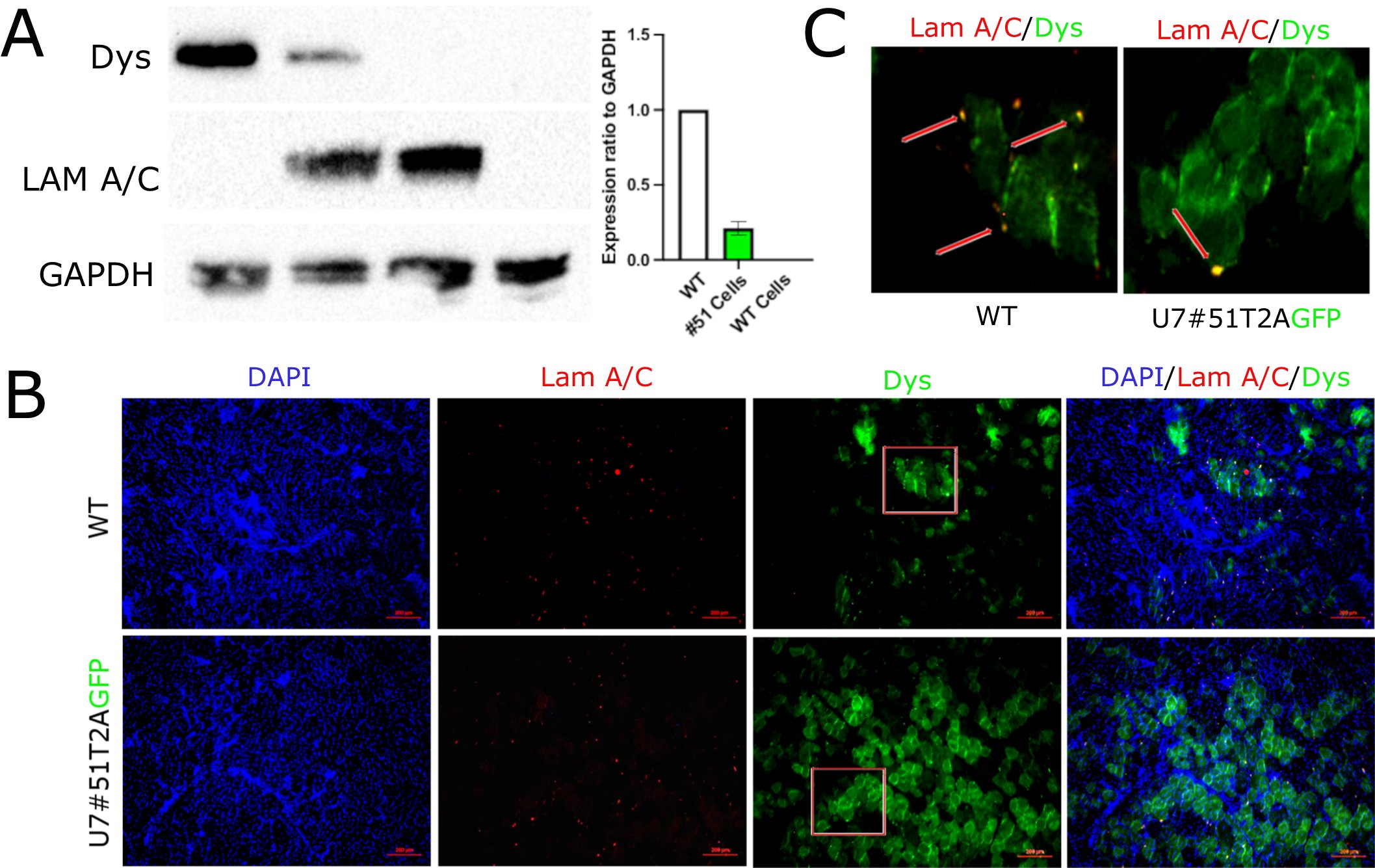
Authors discuss that this works shows that the approach may work and that currently a trial with local injections in Duchenne patients is ongoing and that a follow-up trial with intraarterial injections may follow. Authors do stress the efficiency in mice is rather low.
Authors do not discuss the potential risk of the lentiviral vector integrating in parts of the DNA where you do not want it to be integrating (e.g. activating an oncogene). Authors outline that levels were therapeutic – without adding specifically this was in a mouse model.
We know mice have better quality muscle so the situation in the human will be different with more active pathology, more fibrosis and adiposis and of course a much larger scale (the largest mouse muscle is about 1 gram…). These caveats are not discussed.
I hope the results from the clinical trials will be more positive. However, I worry that they will be disappointing for all the reasons given. One last concern is the number of cells that will be needed when we upscale from mouse to humans. To be continued.

