

#apaperaday: Adeno-Associated Virus (AAV)-Mediated Gene Therapy for Duchenne Muscular Dystrophy: The Issue of Transgene Persistence
In today’s #apaperaday, Prof. Aartsma-Rus reads and comments on the paper titled: Adeno-Associated Virus (AAV)-Mediated Gene Therapy for Duchenne Muscular Dystrophy: The Issue of Transgene Persistence.
Today’s pick is on AAV gene therapy persistence for DMD published by Manini et al in Frontiers in Neurology. Selected because today the Italian International Conference on Duchenne and Becker Muscular Dystrophy has a session on gene therapy. Doi 10.3389/neur.2021.814174.
Duchenne is caused by lack of dystrophin. Gene therapy to deliver a dystrophin gene is being explored. The review paper covers the AAV virus used for this, preclinical and clinical work done and has a focus on the persistence of the micro-dystrophin.
The best option to deliver a copy of a gene (transgene) is currently a viral vector. This is a virus from which all virus genes are removed and the copy of the transgene is inserted with a promoter (volume switch) so the transgene is expressed where it should be expressed.
For Duchenne the AAV virus has been used as this is the only virus that can deliver to muscle and heart. The preference of a virus is called tropism. Selecting a virus with a tropism for muscle means lower doses are needed to get sufficient amounts in muscle.
Authors outline the biology of AAV – a small parvovirus (20-28 nanometer, 1 nanometer is a millimeter divided by a million!) and it is a dependovirus – meaning it needs other viruses to replicate (for transgene delivery it cannot replicate even then – gene required are removed).
AAV genes or transgenes do not integrate in human DNA but persist in the cell where they are used to produce virus proteins (for normal AAV), or the protein of choice (for gene therapy – micro-dystrophin in case of Duchenne). When cells divide transgenes will dilute and be lost.
While AAV has a tropism for muscle, its primary target is the liver. Different types of AAV (serotypes) have different affinity for skeletal muscle and heart. AAV9 and AAV74 are currently the best options and used in gene therapy trials for Duchenne.
AAV does not cause a disease in humans. However, it does trigger an immune response. This means that people who have encountered AAV before, cannot have AAV gene therapy. It also means that for gene therapy immunosuppression is needed.
AAV gene therapy for muscle diseases needs huge amounts of viruses despite the tropism. This is because we have a LOT of muscle (~30-40% of our body mass). Liver damage has been observed (since a lot of virus go to the liver). Sadly also AAV treated patients have died.
4 patients with X-linked myotubular myopathy have died. These all had preexisting liver pathology. In Duchenne patients AAV treatment has resulted in severe side effects including kidney injury, thrombocytopenia (reduced platelets) and sadly also 1 death.
This underlines that AAV gene therapy is not without risks. Research is ongoing to try and mitigate the risks and the side effects and to e.g. be able to predict why some patients have no or little side effects, while others have severe side effects using the same dose.
Because AAV is so small it cannot accommodate much DNA, only ~4.700 base pairs. The protein code for dystrophin is much larger. Therefore a micro-dystrophin is needed, containing the bare essential dystrophin domains. This – with a muscle/heart volume switch (promotor) fits in AAV.
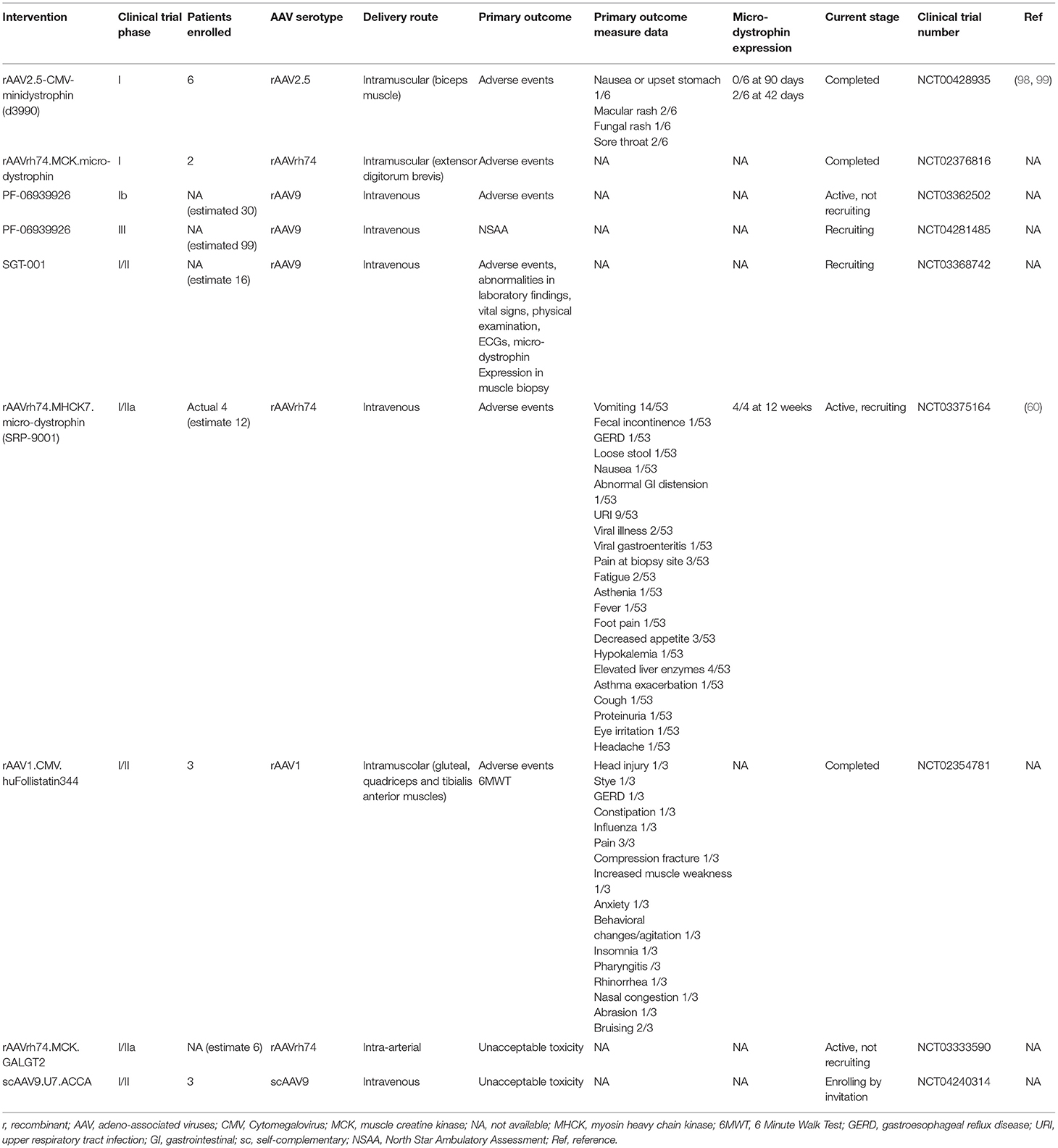
Authors outline the optimization of micro-dystrophin, AAV serotypes for muscle & heart delivery using preclinical studies in mouse & dog models. They also outline the clinical trials ongoing for Sarepta/Roche, Pfizer and Solid (see table 2 for overview).
Note that Genethon is also conducting a clinical trial (not listed in the table). Authors also discuss AAV gene therapy for Duchenne using other transgenes, those that should improve muscle quality (e.g. utrophin, follistatin and GALG2).
I think there is more merit in replacing missing protein than trying to upregulate existing proteins with gene therapy – there might be other ways to upregulate proteins that do not involve AAV precluding patients from AAV to replace missing protein due to immune response.
Finally the focus is on the micro-dystrophin persistence. Normally gene therapy is seen as a 1 time treatment. However, micro-dystrophin is not fully functional so with time the skeletal muscle will be damaged – during this process micro-dystrophin and transgenes will be lost.
So far studies on persistence have primarily been done in mice, where long term (lifelong) is 2-3 years, which does not really provide answers for humans. In healthy muscle transgene expression is long, but of course even with micro-dystrophin Duchenne muscle will not be healthy.
Since there is no turnover in heart, the expectation is that transgene persistence will be longer there. There is no answer yet on how long micro-dystrophins will persist. However, studies to see whether repeat administration is feasible are ongoing in animal models.
I think it is good that people are preparing in case re-administration is needed. However, what is not mentioned in the paper is that we currently do not know if micro-dystrophin is even functional in humans. The fact that it is in 4-legged animals does not mean it will be for us.
Pictures by Annemieke, used with permission.


{kind=link}